The EU’s MDR and IVDR impose evolving compliance demands through 2026 and beyond, creating significant challenges for MedTech procurement teams. Mandatory EUDAMED rollout, supply interruption obligations, and regulatory reforms intensify supply chain risk and cost pressure. From May 2026, EUDAMED will expand data submission and transparency requirements. Extended IVDR transitions through 2026-2029 reduce shortage risks but require strict tracking of notified body agreements. To mitigate risk, procurement must embed regulatory milestones into sourcing, compliance dashboards, and early notified body engagement.[3][2]
Industry Landscape & Regulatory Shifts
EU MDR/IVDR enhance safety and traceability but raise compliance complexity, especially for SMEs, due to limited notified bodies. Amendments extend IVDR timelines and require supply interruption notices. From 28 May 2026, key EUDAMED modules includes: Actor Registration, UDI/device registration, NB certificates, and market surveillance become mandatory, increasing data transparency and reporting. [3][2]
All suppliers must obtain a Single Registration Number (SRN), making regulatory compliance a procurement prerequisite. This elevates supplier risk, certification bottlenecks, and cost pressure, demanding proactive, risk-based sourcing strategies to avoid delisting and disruptions. [3][2][10]
This whitepaper aims to provide MedTech companies particularly procurement, supply chain, and regulatory functions with a forward-looking, actionable framework for navigating MDR and IVDR challenges from 2026 onward.
Implementation of Unique IDevice Identification Roadmap:
The implementation of the Unique Device Identification (UDI) roadmap under EU MDR and IVDR marks a significant regulatory shift for procurement functions. UDI rollout through EUDAMED increases procurement complexity and total landed costs due to expanded traceability, labeling updates, clinical documentation, post-market surveillance, and mandatory database registrations.
From 28 May 2026, all new MDR/IVDR devices must be registered in the UDI module, while legacy devices must transition by 27 November 2027; vigilance reporting becomes mandatory from Q2 2027. UDI-DI and UDI-PI codes must follow global standards (GS1, HIBCC, ICCBBA) and integrate into ERP, labeling, and supplier systems. These requirements elevate supplier qualification effort, system integration costs, and pricing pressure, making early supplier readiness and digital harmonization critical for procurement. [3][2][10]
Transition Periods Under MDR & IVDR:
| Regulation | Device/ IVD Class | Transition End Date | Supplier Impact | Procurement Action |
| MDR | Class III & IIb Implantable | Dec 31, 2027 | Certification lapses risk supply continuity | Verify NB status; plan dual sourcing for critical devices |
| MDR | Class IIb, IIa, Class I Sterile/Measuring | Dec 31, 2028 | Low-risk devices still require NB involvement | Ensure written agreements with NBs; schedule audits |
| MDR | Low-risk devices requiring NB involvement | Dec 31, 2028 | Risk of delayed conformity assessment | Confirm supplier declarations; align with NB schedules; implement contingency sourcing |
| IVDR | Class D High Risk (HIV, Hepatitis tests) | Dec 31, 2027 | High-risk IVD suppliers must meet certification | Track contract certification status; escalate delays |
| IVDR | Class C High/Moderate Risk (e.g., cancer tests) | Dec 31, 2028 | Moderate risk; NB concern required | Confirm supplier NB agreements; prepare backup suppliers |
| IVDR | Class B & Class A Low-risk Sterile IVDs (e.g., blood collection tubes) | Dec 31, 2029 | Risk of delay on certificate expiry | Plan requalification if needed; maintain proactive supplier communication |
Sources: “EU Medical Devices Legislation: What You Need To Know Given …,” Arnold & Porter, 2025.
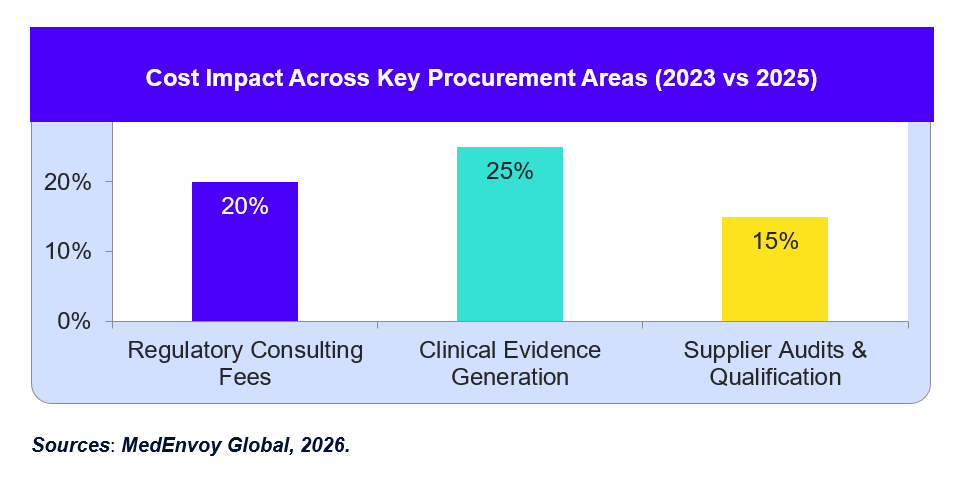

Sources: “EU Medical Devices Legislation: What You Need To Know Given …,” Arnold & Porter, 2025.
List of product lines that may require switching to Notified-Body-Approved suppliers or components due to Reclassification:
Under EU MDR and IVDR, Notified Bodies (NBs) are essential for conformity assessment of medium- and high-risk medical devices and most IVDs, validating regulatory, safety, and performance compliance required for EU market access. MDR classifies devices from Class I (low risk) to Class III (high risk), while IVDR categorizes IVDs from Class A to Class D based on patient and public health risk.
Regulatory reclassification has expanded the scope of products requiring NB-approved suppliers or components. Impacted product lines include reclassified medical devices and IVDs, drug-device combination products, certain PPE, and products undergoing significant design or intended-use changes. These products must undergo NB review to demonstrate compliance with MDR General Safety and Performance Requirements or IVDR scientific validity, analytical, and clinical performance standards.[11]
IVDR additionally mandates Performance Evaluation Plans, Performance Evaluation Reports, and Post-Market Performance Follow-up. Certification obligations extend across manufacturers, OEMs, importers, and distributors. Given documentation intensity, NB capacity constraints, and interpretation variability, early NB engagement is critical to avoid certification delays and supply disruption. [3][2][10][11]
Implementation Guide for Regulatory Transition:
Procurement teams must act decisively to manage MDR and IVDR transition risks. Priority actions include deploying supplier regulatory maturity scorecards, updating contracts with notification, audit, and alternate supply clauses, and segmenting portfolios by regulatory risk.
High-risk SKUs should be dual-sourced or buffered to offset certification delays. Teams must also track EU regulatory reforms, engage in industry consultations, and invest in regulatory automation and analytics. The 2026–2027 period is pivotal; delayed action risks supply disruption, cost escalation, and market withdrawal.[11] [8][2]
Conclusion:
From 2026 onward, increasing MDR/IVDR complexity, NB capacity constraints, and ongoing regulatory reform create a high-risk environment for MedTech companies. For procurement, the necessity is to shift from reactive compliance management to structured, risk-based sourcing. Certification bottlenecks, new obligations such as Article 10a, and supplier compliance gaps heighten the risk of device de-listing. A coordinated approach along with supplier maturity scoring, strengthened contracts, and selective buffer or dual sourcing supports resilience, protects market access, and transforms regulatory pressure into a strategic procurement advantage.
References
[1] “New Regulations – Medical Devices – Public Health – European Union,” European Commission, 2025.
[2] “EU Medical Devices Legislation: What You Need To Know Given …,” Arnold & Porter, 2025.
[3] “New MDR and IVDR regulations: This will change in 2025,” Aristo Group, 2025.
[5] “MDR and IVDR extension & implementation updates,” MDLaw, 2025.
[7] “Industry groups call for significant reforms to MDR, IVDR,” RAPS, 2025.
[8] “EU set to upgrade medtech and diagnostics regulations,” Osborne Clarke, 2025. .
[9] “What’s the Cost of Medical Device Approval in Europe?,” MedEnvoy Global, 2026.
[10] “EUDAMED Mandatory Timelines for MDR and IVDR in 2026,” MDx CRO, Nov. 27, 2025.
[11] MedFilesGroup, “What to know about medical device and IVD class changes and transition periods,” MedFilesGroup, 2026.
Author
S. Akila
Nitin Khaladkar
Related Reading

16 Mar, 2026
The New Tariff Reality: How Global Trade Shifts Are Rewriting Electrical Equipment Sourcing